
治験管理・支援システムのイベント情報
2025年12月以降に開催される治験管理・支援システムのイベント情報を掲載しています。
| イベント名称 | ファーマIT&デジタルヘルス エキスポ 2026 |
|---|---|
| 開催日時 | 2026年4月21日(火)~23日(木) 10:00~17:00 |
| 会場所在地 | 東京ビッグサイト 東8ホール |
| 主催 | インフォーマ マーケッツ ジャパン株式会社 |
| 後援団体 | 日本製薬工業協会、(一社)日本CRO協会、(一社)バイオ産業情報化コンソーシアム、(一社)LINK-J、(一社)日本クラウドセキュリティアライアンス、(一社)ヘルスケアイノベーション協会、(一社)生成AI活用普及協会、(一社)製造DX協会、(一社)HMIC ほか |
| 来場者数 | 約9,300名(2025年実績) |
| 出展対象 | 製薬メーカーの研究・開発・IT・セールス部門、CRO、バイオベンチャー、創薬企業など |
| 公式サイト | https://www.pharmait-expo.com/ |
| イベント名称 | 日本臨床試験学会 第17回学術集会総会 |
|---|---|
| 開催日時 | 2026年2月19日(木)~21日(土) |
| 会場所在地 | 神戸国際会議場(兵庫県神戸市) |
| 主催 | 日本臨床試験学会 |
| 会長 | 真田昌爾(神戸大学医学部附属病院 臨床研究推進センター) |
| 公式サイト | https://jsctr17kobe.umin.jp/ |
| イベント名称 | 日本臨床薬理学会 第46回・第47回学術総会 |
|---|---|
| 第46回 開催日時 | 2025年12月5日(金)~6日(土) |
| 第46回 会場所在地 | ステーションコンファレンス東京(東京都千代田区) |
| 第47回 開催日時 | 2026年11月27日(金)~28日(土) |
| 第47回 会場所在地 | ハピリンホール・アオッサ福井(福井県福井市) |
| 主催 | 日本臨床薬理学会 |
| 公式サイト | https://www.jscpt.jp/meeting |
治験・臨床研究のサポートとして
ニーズの高い支援システムは3種
治験や臨床研究の支援システムを導入する際には、目的に応じて必要な機能を多く搭載したシステムを選ぶことが重要です。
ただし、さまざまな機能を幅広くサポートするシステムは便利な反面、機能の多さに比例して導入や運用コストが増大する傾向があります。
また、機能が過剰に多い場合、利用者が操作に戸惑い、現場での混乱を招くケースも見受けられます。
一方で、「モニタリング業務支援」や「文書管理支援」など、特定の機能に特化したシステムは、目的に応じた機能をシンプルかつ使いやすい形で提供しているものが多く、ITに不慣れな利用者でもスムーズに運用できる点が魅力です。
特に、クラウドベースのシステムは、初期導入コストを抑えつつ、場所を問わずアクセスできる柔軟性や、自動アップデートによる最新機能の活用が可能なため、利用者の負担軽減や満足度向上に大きく寄与します。
現在、支援システムとしてニーズが高まっている機能は、大きく次の3つです。

モニタリング業務の煩雑な手間を減らしてくれるシステム。
例えばこんな機能
- リアルタイムでデータ収集の進捗・逸脱を確認できる
- 各施設・各患者の状況が一目でわかる
- モニタリング報告書がほぼ自動的に作成される

入手したデータを元に、各書式に応じた文書を作成するシステム。
例えばこんな機能
- 各ガイドラインに沿った文書をほぼ自動で作成する
- 電子署名や版管理機能などがあり文書をデータ上で管理できる
- PDFやExcelで出力できる

電子的な症例報告書を作成し、ネット経由でサーバに取り込むシステム。
例えばこんな機能
- 電子症例報告書(eCRF)の作成や既存データの転記ができる
- 患者への説明や承諾を得る作業が同一システム内で完結できる
おすすめの治験管理
・支援システム
(CTMS)
【機能別】
おすすめシステム3選
被験者管理や進捗確認、文書対応、監査準備など、治験業務に伴う煩雑な作業を支援し、効率化と法規制対応を実現する治験支援システム。 ここでは、モニタリング・文書管理・EDCの主要機能別に、実績と信頼性のあるおすすめ3システムをご紹介します。
(治験の実施状況確認・報告)
(SOLUMINA)
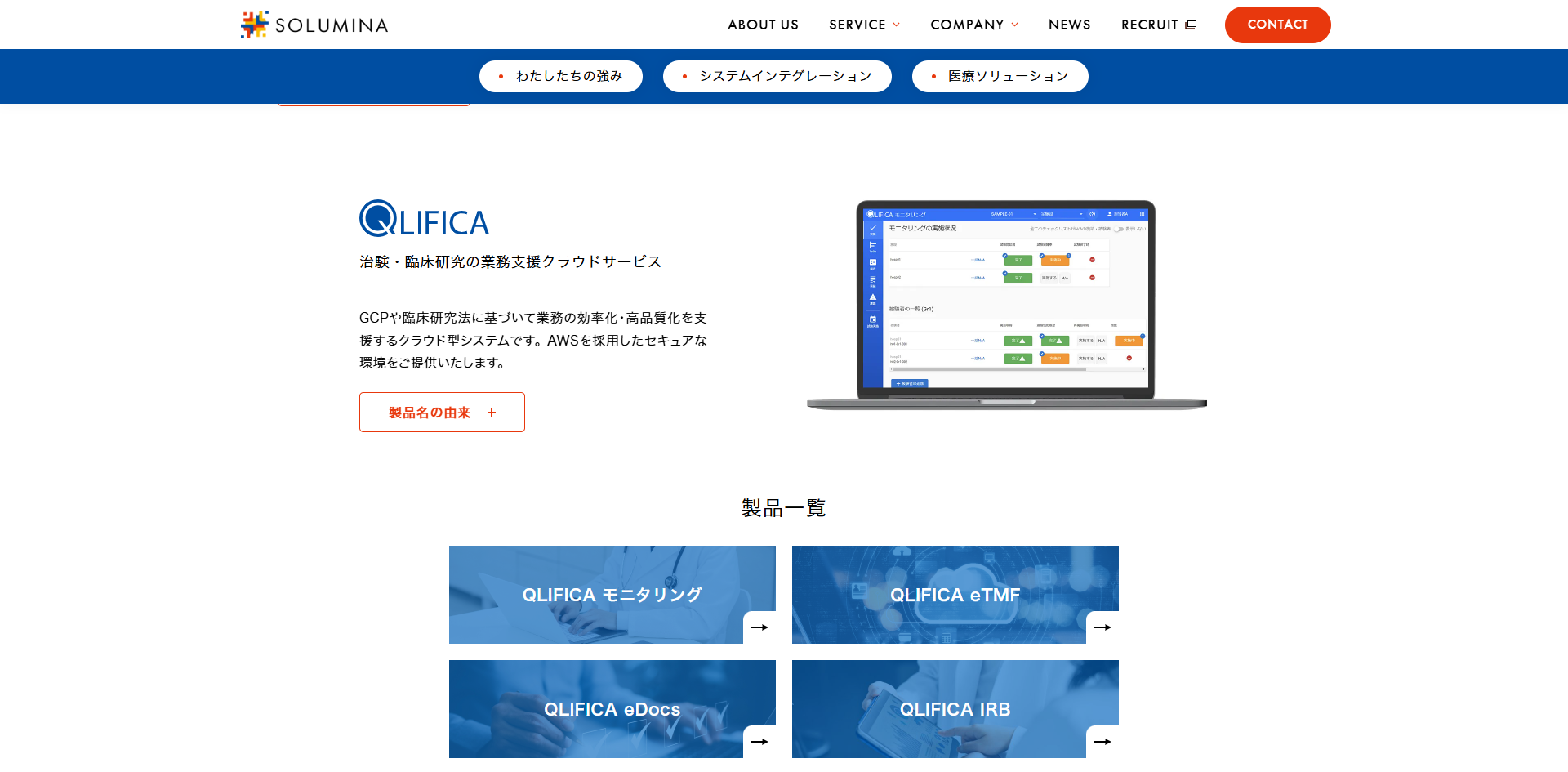
画像引用元:SOLUMINA 公式HP(https://solumina.co.jp/service/#qlifica)
- 施設・症例・CRA単位で進捗をリアルタイムに見える化
- チェック内容から報告書を自動作成、承認も一括完了
- 課題の対応状況を履歴付きで一元管理、監査対応も容易
- 複数施設の進捗と履歴を即時に把握し、作業漏れを防ぐ。
- 文書・IRB・監査対応まで一括管理し、全体業務を効率化。
(治験関連文書の保管・共有)
(Agatha)

画像引用元:Agatha 公式HP(https://www.agathalife.com/)
- 文書ごとの承認状況をリアルタイムで一元管理
- 電子原本として保管し、法規制や監査に対応
- 試験や組織単位で柔軟に文書構成を設計・運用可能
- 契約書や申請書類の承認・版管理を統一し、整合性と履歴を正確に管理
- 原本性を保った電子保管で、GCP・ER/ES対応を文書単位で実現
(電子症例報告)
(メビックス)

画像引用元:メビックス 公式HP(https://www2.mebix.co.jp/services/edc/)
- 入力内容に応じて画面項目を自動制御
- 入力時に整合性エラーを即時にチェックして通知
- クエリ対応履歴を一覧表示し進捗を共有
- 入力作業がスムーズになり、記入ミスや作業ストレスを減らせる
- DMや統計担当者とのやりとりが明確になり、確認・集計の手戻りがなくなる
おすすめ治験管理・支援システム(CTMS)3選をもっと詳しく
(治験の実施状況確認・報告)
QLIFICA(クリフィカ)
モニタリング
(https://solumina.co.jp/service/#qlifica)
QLIFICA モニタリング
| 金額 | 【企業向け】 ライセンス数無制限:年間253万円(税込) /ライセンス50:年間154万円(税込) /ライセンス20:年間90.2万円(税込) 【大学病院向け(キャンペーン価格)】 ・ライセンス30:年間66万円(税込)※容量制限あり(200GB) |
|---|---|
| 構築期間目安 | 要問合せ |
搭載されている機能
- リアルタイムで試験の進捗情報を収集できる
- 発生した課題を一覧で確認できる
- チームで情報共有できる
- チェックリストを読み込みモニタリング実績を登録できる
- 対応漏れ防止にアラート機能を搭載
- モニタリングの実績に関しては、経時的にExcelの出力が可能
- モニタリング報告書は、実施記録からほぼ自動的に作成可能
- モニタリングの実施記録を、チェック項目に回答するだけで登録できる
- モニタリング報告書に関する資料は、簡単にファイル添付可能
- Excelのまま設定書をアップロードできる
…など
QLIFICA モニタリングの特徴
報告作業を効率化する
操作性と記録支援
治験や臨床試験におけるモニタリング業務を効率化するクラウド型支援システムです。 チェックリストの取り込み機能により、Excelで作成した設定書のアップロードが可能で、規制や研究ごとの進捗・逸脱管理に対応します。
実施記録はチェック項目への回答形式で入力でき、報告書もほぼ自動生成されるため、記録負担の軽減とヒューマンエラーの防止を両立します。 操作性に優れたUIも特長です。
QLIFICA eTMF
| 金額 | 【企業向け】 基本料金:年間253万円(税込)+初期費用121万円~(税込)/ライセンス料:年間5.5万円(税込) 【医療機関向け】 ・試験数10:年間66万円(税込)/試験数30:年間92.4万円(税込)/試験数50:年間118.8万円(税込) ・環境構築費用:38.5万円~(税込) |
|---|---|
| 構築期間目安 | 要問合せ |
搭載されている機能
- DIA TMF Reference Modelに基づいた文書分類設定が可能
- 文書のワークフロー管理により、レビューから承認までのプロセスを統制
- 文書のバージョン管理と固定機能で、改訂履歴の追跡と誤操作の防止が可能
- 文書情報を条件で検索し、結果をExcel形式でダウンロード可能
- 試験ごとの文書を一括でダウンロードできる機能を搭載
- QLIFICAモニタリングと連携し、報告書を自動で格納
- ユーザーごとに表示言語(日⇔英)の切り替えが可能 他
QLIFICA eTMF の特徴
文書管理を効率化する
クラウド型eTMF
治験や臨床研究に関わる文書情報をオンラインで一元管理するクラウドサービスです。 DIA TMF Reference Modelなどの分類構成を試験単位で適用し、文書の集約・整理・ワークフロー管理・ファイル保管を実現します。
モニタリング報告書は自動で格納され、チェックリストと文書の紐づけも可能です。 高セキュリティ環境での保管や操作ログの記録により、文書管理の効率化と品質向上を支援します。
QLIFICA eDocs
| 金額 | ・基本利用料金(年間):88万円(税込) ・ユーザーライセンス料(年間/1ライセンス):2.64万円(税込) ・初期費用(環境構築+操作レクチャー):23.1万円(税込) ・容量追加オプション(年間):6.6万円(税込)/100GB単位 【医療機関向け】 ・ライセンス30(年間):22万円(税込) ・初期費用:22万円(税込) ・容量追加オプション(年間):6.6万円(税込)/100GB単位 |
|---|---|
| 構築期間目安 | 要問合せ |
搭載されている機能
- 任意のフォルダ構成で文書を管理する設定が可能
- 文書情報にタグを付与し、検索性を向上
- 文書のワークフロー管理により、レビューから承認までのプロセスを統制
- 文書のバージョン管理と固定機能で、改訂履歴の追跡と誤操作の防止が可能
- 文書情報を条件で検索し、結果をExcel形式でダウンロード可能
- 試験単位で文書を一括ダウンロードできる機能を搭載
- ユーザーごとに表示言語(日⇔英)の切り替えが可能 他
QLIFICA eDocs の特徴
手軽に始められるミニマルな
文書管理
文書情報の電子化を手軽に実現するクラウドサービスです。 フォルダ構成でのファイル管理やレビュー・承認等のワークフロー管理、バージョン管理などの基本機能を備えています。
ER/ES指針に対応した電子原本管理や監査証跡の記録も可能で、高セキュリティ環境で文書を保管・共有できます。 ミニマルな機能で、初めての文書管理システム導入にも適しています。
QLIFICA IRB
| 金額 | 要問合せ |
|---|---|
| 構築期間目安 | 要問合せ |
搭載されている機能
- 委員会の概要情報や構成メンバーをシステム上で管理
- 医師主導治験・企業治験など各種治験情報を一元で取り扱い
- 治験依頼書(書式3)の作成と関連資料の添付に対応
- 治験審査依頼書(書式4)の作成と資料一式の提出に対応
- IRB会議の開催予定、委員の出欠管理、進捗記録を統合管理
- 委員による資料の閲覧・ダウンロード、閲覧済ステータスの確認が可能
- 審査結果通知書(書式5)を作成し、依頼者・医療機関への提出まで対応 他
QLIFICA IRB の特徴
治験審査業務を支援する
クラウドサービス
QLIFICA IRBは、治験審査に関する業務全般を支援するクラウドサービスです。審査の依頼・会議の開催・審査結果の通知までを一元管理し、統一書式(書式3、4、5、参考書式1)の効率的な作成を支援します。
IRB資料はセキュアな環境で安全に保管・共有でき、ペーパーレス化を推進します。院内IRBだけでなく、中央IRBの運用にも対応しています。
QLIFICAを提供する
SOLUMINAについて
QLIFICAシリーズを提供する株式会社SOLUMINAは、治験・臨床研究業務の効率化と品質向上を支援するクラウドサービスの開発に特化した企業です。臨床試験分野に精通した体制を持ち、GCPやER/ES指針に準拠した信頼性の高いソリューションを展開しています。
モニタリング、文書管理、IRB対応など、各機能を独立または連携して活用できる点が強みであり、製薬企業、CRO、医療機関など多様な導入先に対応可能です。
QLIFICA モニタリングを
提供するSOLUMINAの会社情報
| 会社名 | 株式会社SOLUMINA |
|---|---|
| 所在地 | 神奈川県横浜市西区みなとみらい4-4-2 横浜ブルーアベニュー13階 |
| 電話番号 | 045-211-0761 |
| 許認可 | Pマーク(2006年取得)、ISO27001(2012年取得) |
(治験関連文書の保管・共有)
Agatha(Agatha)
(https://www.agathalife.com/)
Agatha Basic
| 金額 | 要問合せ |
|---|---|
| 構築期間目安 | デフォルト設定の場合は約1か月、バリデーション対応時は約3か月 |
搭載されている機能
- 文書の登録・編集・共有をクラウド上で管理
- 文書タイプや属性を用いたメタデータ管理に対応
- レビュー・承認のワークフローを標準搭載
- 版管理と監査証跡の自動記録で改ざんを防止
- 電子署名機能で正式な文書承認を実現
- 日本語・英語の切り替えにより多施設展開に対応
- 柔軟な検索・抽出・分類機能で文書を一括管理 他
Agatha Basic の特徴
シンプルかつ確実に文書管理を
始められる
治験・臨床研究に関する文書の保管・共有・承認を、クラウド上で手軽に始められるエントリーモデルです。複雑な設定を必要とせず、標準テンプレートの利用でスムーズな導入が可能。
電子署名や監査証跡、ワークフロー機能など、GCPやER/ESに求められる基本要件を標準装備しており、必要十分な機能性と高い操作性を両立しています。柔軟なアクセス権限や言語切替にも対応しており、少人数体制や初期導入に適した構成です。
Agatha 施設文書保管+IRB
| 金額 | 要問合せ |
|---|---|
| 構築期間目安 | シンプル構成で1~2か月、CSV実施時は約3か月 |
搭載されている機能
- 治験関連文書を電磁的に授受・保管し一元管理
- 試験ごとにワークスペースを分けて施設単位で文書整理
- IRB申請書類の作成から履歴管理までクラウド上で対応
- 版管理・監査証跡の自動記録により改訂履歴を可視化
- レビュー・承認のワークフローと電子署名に標準対応
- 外部共有時も制御できる柔軟なアクセス権限設定
- 日本語・英語対応により多施設・多国対応が可能 他
Agatha 施設文書保管+IRB の特徴
IRB申請業務と文書管理を
ひとつの画面で完結
医療機関における治験関連文書の保管とIRB業務の電子化を同時に支援するクラウドサービスです。試験ごとのワークスペース管理により、申請書・同意書・審査資料などを一括で整理・共有可能。
IRB申請の流れもシステム上で完結し、進捗状況や文書履歴を関係者間で即時に把握できます。 電子署名、監査証跡、アクセス制御なども備え、厚労省・PMDA対応や多施設運用に求められる要件をしっかりとカバーします。
Agatha 施設文書保管
+IRB の機能を
公式サイトでもっと詳しく
Agatha eTMF
| 金額 | 要問合せ |
|---|---|
| 構築期間目安 | 標準テンプレート導入時は2~3か月、追加設定がある場合は3か月以上(要件により変動) |
搭載されている機能
- DIA TMF参照モデルに基づいたフォルダ・文書構成を提供
- 文書の進捗状況をダッシュボードで可視化
- 監査官専用の閲覧権限設定で監査・査察対応を支援
- 文書のドラフト作成から確定、QCチェックまで一元管理
- 外注先やユーザーごとに柔軟な操作権限を設定可能
- 文書をクラウド上に一元保管し、施設・試験単位で整理
- ER/ES指針、FDA 21 CFR Part 11に準拠した信頼性保証体制を構築 他
Agatha eTMF の特徴
文書の整備と監査対応を
両立するクラウド型eTMF
治験業務に欠かせない必須文書・関連文書を、プロジェクト単位で一元的に管理できるクラウド型eTMFシステムです。 DIA TMF参照モデルに準拠したフォルダ構成をもとに、文書の分類・保存・進捗確認までを体系化。ドラフト作成からQCチェック、最終承認、バージョン管理までの一連の流れを効率よく処理できます。
さらに、監査官専用の閲覧権限を設定でき、操作履歴もすべて記録。ER/ES指針および21 CFR Part 11にも準拠しており、国内外を問わず、品質管理・監査対応に強い体制構築を支援します。
Agathaを提供する
アガサ株式会社について
Agathaシリーズを提供するアガサ株式会社は、治験・臨床研究における文書管理業務の効率化を目的としたクラウドサービスの開発・提供を行うヘルステック企業です。 GCPやER/ES指針、21 CFR Part 11への準拠を前提とした高セキュリティ設計により、医療機関・製薬企業・CROなどで幅広く導入が進んでいます。
IRB申請やeTMF運用、文書の電子保存まで、導入しやすく直感的に使えるUIと柔軟な運用性を兼ね備えており、国内外の臨床現場に適応する実績あるサービスを提供しています。
Agatha CTMSを提供する
アガサの会社情報
| 会社名 | アガサ株式会社 |
|---|---|
| 所在地 | 東京都中央区日本橋兜町7-1 Kabuto One 9階 WeWork |
| 電話番号 | 050-3188-5299 |
| 許認可 | ISO27001(2018年取得)、ISO9001(2021年取得) |
(電子症例報告)
CapTool® シリーズ
(メビックス)
(https://www2.mebix.co.jp/services/edc/)
割付君
| 金額 | 初期費用:55万円~(税込) 月額運用費:2.2万円~(税込) |
|---|---|
| 構築期間目安 | 最短2週間程度(設定内容により変動) |
搭載されている機能
- 症例登録および割付に対応(最小化法・置換ブロック法)
- ロジカルチェックで入力ミスを自動検出できる(オプション)
- 症例の修正や削除を管理画面から実施できる
- 症例登録完了時に通知メールを自動送信できる
- 症例一覧をCSV形式で出力・ダウンロードできる
- 担当者ごとのID/パスワードでアクセス管理できる
- 登録・操作履歴を監査証跡として記録できる
割付君 の特徴
使いやすさに特化したWeb型割付
支援システム
症例登録と割付に特化したWebベースの簡易ツールです。最小化法や置換ブロック法に対応しながら、最短2週間でのスピード導入が可能。
医師やCRCの実務に即したシンプルな画面設計で、初めて割付システムを導入する医療機関や研究者にも適しています。ロジカルチェックやデータ修正、監査証跡などの基本機能も備えており、短期研究や少数症例での活用にも適した構成です。
CapTool Lite
| 金額 | 初期費用:77万円~(税込) 月額運用費:3.3万円~(税込) |
|---|---|
| 構築期間目安 | 最短2週間程度(設定内容により変動) |
搭載されている機能
- 症例登録および割付を画面入力で簡単に行える
- 追跡調査用の入力項目を症例単位で設定できる
- 繰返し画面・繰返し項目の入力管理に対応
- ロジカルチェックで入力不備を検出できる(オプション)
- 症例データの修正・削除を管理側でコントロールできる
- 症例一覧をExcel形式で出力・保存できる
- 監査証跡を自動で記録し、確認・証跡管理に対応できる
CapTool Lite の特徴
中規模研究向けに機能を絞った
シンプルEDC
割付・症例登録から簡易な追跡調査までをカバーするエントリーEDCです。必要な機能を厳選し、使いやすさとコストパフォーマンスを両立。 ユーザー50名・300症例規模を想定した料金設計に加え、アカデミックライセンスにも対応しています。
追跡機能やロジカルチェック、データ修正、監査証跡といった品質確保機能を備え、EDCを初めて導入する施設やCROに適した構成です。
CapTool Cloud
| 金額 | 要問合せ |
|---|---|
| 構築期間目安 | 要問合せ |
搭載されている機能
- 症例登録・追跡・割付をクラウド上で一元管理できる
- 動的画面制御により項目表示を条件に応じて切替できる
- クエリの発行・対応履歴を記録して管理できる(予定)
- 繰返し入力・繰返し項目に対応した入力画面を構築できる
- 登録完了時に自動で通知メールを送信できる
- ID・パスワードごとに操作権限を制御できる
- 監査証跡・操作履歴をシステム上で保持できる
CapTool Cloud の特徴
柔軟性と拡張性を備えた
次世代クラウドEDC
EDCの運用をクラウド上で完結させる拡張型の電子データ収集システムです。症例登録、追跡調査、画面制御、クエリ管理、コーディング(近日対応)など多機能に対応しながら、案件ごとに柔軟なカスタマイズが可能。
操作権限や監査証跡、通知設定も細かく制御でき、EDCを内製したいCROや機能統合を求める製薬企業にとって有力な選択肢となります。
CapTool Cloud の機能を
公式サイトでもっと詳しく
CapTool mini
| 金額 | ・初期費用:400万円~(税込) ・月額運用費:20万円~(税込) |
|---|---|
| 構築期間目安 | 要問合せ(導入内容により変動) |
搭載されている機能
- 症例の進捗や入力状況をステータスで可視化できる
- クエリを介した質問・指摘事項の記録・対応ができる
- 繰返し項目・画面の入力処理を柔軟に設定できる
- ロジカルチェックや脱落後入力制御に対応している
- 電子署名機能で記録内容の承認・確定を行える
- 監査証跡で修正・操作履歴を一括管理できる
- 症例データをCSVでダウンロードして集計可能
CapTool mini の特徴
軽量かつ本格的な機能を備えた
導入型EDC
小規模~中規模試験に特化したミドルレンジのEDC製品です。 CapTool Primeに近い入力管理機能を持ちながら、導入コストを抑えた構成が特長。症例ステータスの可視化、クエリ管理、電子署名、コーディング対応など、GCP準拠の試験に必要な機能を一通り備えています。
CROや中小製薬企業が、より実務的かつ効率的にEDCを運用したい際の選択肢として適しています。
CapTool Prime
| 金額 | ・初期費用:600万円~(税込) ・月額運用費:20万円~(税込) |
|---|---|
| 構築期間目安 | 要問合せ(導入内容により変動) |
搭載されている機能
- 症例登録・割付を多様な方式で運用できる(最小化法等)
- 症例ごとの進捗状況やステータスを一覧で確認できる
- クエリの発行・回答履歴を記録し、確認可能にする
- ロジカルチェック・画面制御・項目制御を統合管理できる
- 電子署名により記録内容の法的な承認を実施できる
- 監査証跡で全操作の履歴を自動的に記録できる
- コーディング機能で用語統一・辞書管理に対応している
CapTool Prime の特徴
製薬企業・グローバル対応を想定した
最上位モデル
フル機能を備えたエンタープライズ向けEDCプラットフォームです。進捗管理、クエリ、ロジカルチェック、電子署名、コーディング機能を標準搭載し、GCP・ER/ES・21 CFR Part 11にも対応。
多施設・多診療科・国際共同研究にも対応できる高い拡張性と堅牢な運用体制を持ち、社内にEDC基盤を構築したい製薬企業や、グローバルスタディを主導するCROに適しています。
CapTool Prime の機能を
公式サイトでもっと詳しく
CapTool® シリーズを提供する
メビックスについて
メビックス株式会社は、臨床試験・医師主導治験・製造販売後調査(PMS)などの多様な臨床研究を支援するCROとして、ITと医療現場の融合を強みに持つ企業です。
EDC開発においては、自社開発の「CapTool®」シリーズを通じて、試験規模やニーズに応じた柔軟なシステム導入を実現。 割付支援からフルスケールEDCまで幅広く展開し、製薬企業や医療機関、CROの業務効率化と品質向上を支えています。
メビックスの会社情報
| 会社名 | メビックス株式会社 |
|---|---|
| 所在地 | 東京都港区虎ノ門3-8-21 虎ノ門33森ビル10階 |
| 電話番号 | 03-4362-4500 |
| 許認可 | 要問合せ |
【目的別】おすすめの治験⽀援
システム
治験のモニター業務は
もっと楽にできる!
導⼊検討時に注⽬すべき機能

治験や臨床研究でもコストのかかるモニタリング業務。データの正確性とデータ収集状況を常時チェックする体制が求められるため、チェックリストを始めとしたツールで厳格に管理しなければならず、現場の負担も増える一方です。
負担を減らすために必要なこと、考えたいことをまとめました。
イベント情報
- 第43回日本臨床薬理学会学術総会in横浜
2022年11月30日~
https://www.congre.co.jp/jpw2022/ - 日本臨床試験学会第14回学術総会in金沢
2023年2月9日~
https://convention.jtbcom.co.jp/jsctr2023/ - ファーマIT&デジタルヘルス エキスポ 2023
2023年4月19日~
https://www.pharmait-expo.com/
治験⽀援システム選びの際に
覚えておきたい⽤語まとめ
| CTMS | 臨床試験管理システム、「Clinical Trial Management System」の略称です。臨床試験のプロセス管理が容易になり、スピーディーな対応を実現できます。 |
|---|---|
| PMDA | 独立行政法人医薬品医療機器総合機構、「Pharmaceuticals and Medical Devices Agency」の略称です。国民保険の向上のために、医薬品や医療機器の品質・有効性・安全性の情報収集や分析をし、安全対策を行っています。 |
| IRB | 治験審査委員会、「Institutional Review Board」の略称です。治験実証前に、治験内容を安全性・倫理性・有効性の観点から審査します。 |
| CSV | コンピュータ化システムバリデーション、「Computerized System Validation」の略称です。コンピュータ化システムについて、開発プロセスの最適性と妥当性の検証・文書化のガイドラインが定められています。 |
| ER/ES指針 | ERは「Electronic Record」で電子データ、ESは「Electronic Signature」で電子署名の略称です。厚生労働省がER/ES指針を示しており、電子書類の信頼性を確保するために要件を満たす必要があります。 ※参照元:[PDF]独立行政法人医薬品医療機器総合機構「治験及び調査における 電磁的記録の利用について」https://www.pmda.go.jp/files/000161516.pdf |
| CRF | 症例報告書、「Case Report Form」の略称です。臨床試験を行った時の検査データと、副作用の情報をまとめた報告書です。 |
| SDV | 直接閲覧、「Source Document Verification」の略称です。モニタリング業務の一環として、記録や報告書の分析をして確認する作業を指しています。 |
| EDC | 臨床試験支援システム、「Electronic Data Capture」の略称です。臨床試験のデータを電子化し、管理するシステムを指しています。 |
治験⽀援システムを
導⼊した研究機関の声
実際に治験の支援システムを導入し、治験業務の効率が上がった・負担が減った企業や大学病院の声をまとめました。
利便性とセキュリティが向上
これまで検査データや症例報告書のやりとりをメールで行っていましたが、メールでのやりとりは非常に煩雑ですし、パスワードをかける手間がかかり、誤送信のリスクもあります。そのため、利便性向上とセキュリティ向上のためにシステムを導入することにしました。(中略)Agathaには版管理機能があり、すべての版が保存されるため、進捗をエクセルに記入して保存するだけで、関係者と常に最新情報を共有し、過去の情報も必要なときに取り出すことができます。(中略)分担医師・責任医師による確認もレビュー機能で行っています。今後は、他の疾患の臨床研究、倫理審査申請、学会内の運営(理事会、議事録等)などにも、順次利用を広げていく構想です。
操作性と汎用性の高いシステムで課題解決
研究者が自らモニタリングを実施している研究において、当グループがモニタリングの進捗状況を確認し、適宜指導を行うこととなりました。その際、効率的にモニタリングの進捗状況を確認する手段を検討し、QLIFICAに辿り着きました。また、当院では治験用の臨床研究業務管理システムを既に導入していますが、当グループは様々な臨床研究を支援しているため、治験だけでなく臨床研究にも適した扱い易い効率的なシステムの導入を行い、効果的な業務推進環境を整える必要がありました。(中略)臨床研究を取り扱いやすく、誰もが利用しやすい操作性が当グループの課題解決に役立つと考えました。
国内で普及しているCTMS・治験支援
システム一覧
CTMSシステム一覧
治験分野で高いシェアを誇るアガサの製品です。クラウドでのモニタリング報告書の作成・管理、Issue登録・管理を行うことができます。
知名度の高い富士通のDdworks21。機能を多面的に備え、大規模な治験にも対応します。
ファーマメディカルソリューションが提供するCTMS。研究者と事務局のやり取りをスムーズに。
MedidataのRave CTMSは、9,000以上のCTMS試験数に使用される治験管理システムです。
※2021年7月調査時点の数字。参照元:MEDIDATA「Rave CTMS」https://www.medidata.com/jp/clinical-trial-products/clinical-operations/ctms
さまざまなソリューションを提供するVeevaのマルチテナント型クラウドソリューションです。
モジュール形式で、後から機能の追加・更新が可能。カスタムしやすいシステムといえるでしょう。
富士通の「HOPE eACReSS(イーアクレス)」は、シンプルなUIでプロジェクト管理を行います。
同じく富士通が提供する「HOPE NMGCP」は、富士通が提供する電子カルテと標準的に連携可能です。
「Clear Trial」はアメリカの大手ソフトウェア開発企業のオラクルが手掛ける治験・臨床研究支援システム。タスクの一覧化からスケジュール・コスト・リソース管理まで、臨床試験に関わ情報を可視化し、業務を支援してくれます。
医療業務支援のシステム開発で実績があるNDDの治験管理システムです。製薬会社が持つ治験情報や実施計画に関する情報と、院内のオーダリングシステムの連携が可能で治験業務の効率を向上を助けてくれます。
独立調査組織はもちろん大規模な病院や学術機関など、臨床試験を行うあらゆる機関で使用できるよう最適化されたシステム治験・臨床研究支援システムです。
臨床開発分野における大手企業の一つであるALMACが手掛ける治験・臨床研究支援システム「ALMAC ART」。転記ミスやデータの欠損などを防ぐ自動応答技術(IRT)「IXRS®3」を拡張機能として導入することも可能です。
モニタリング業務をサポートするシステム
株式会社SOLUMINAの提供する、クラウド型のモニタリング業務サポートシステムです。
文書管理機能で知名度の高い「Agatha」のモニタリング報告書の作成・管理、Issueの登録管理により、臨床試験情報とプロセスを可視化するシステムです。
オレガの「Alternax® MPS」はモニター管理者の管理を容易にするシステムです。
文書管理に特化したシステム
「QLIFICA(クリフィカ)eTMF」は、株式会社SOLUMINAが提供するクラウドサービスです。情報の集約とワークフローのクラウド化により、効率的な文書管理を実現します。
Agathaの提供する「Agatha eTMF」は、文書管理に必要な機能を多々備えたシステムです。
TMFの作成時にリアルタイムに文書を管理する、Veeva社の「Value eTMF」。同社はCMTSなど複数ソリューションを提供します。
MEDIDATAの文書管理システム「Rave eTMF」。Rave CTMSと併用することで試験をスムーズに管理できます。
公益社団法人日本医師会が提供している「カット・ドゥ・スクエア」は無償で使用できるソリューションです。
株式会社ビッグバンの「BIGVAN」は「治験の依頼等に係る統一書式」に統一したフォーマットで文書管理業務をサポート。
EDCに特化したシステム
メビックスの「Cap Tool(キャップツール)」は動的画面制御により操作しやすいUIを提供します。
Veevaが提供するEDCシステム「Vault EDC」。アジャイルデザインアプローチで構築期間短縮を図ります。
メハーゲングループが提供する、臨床試験支援のためのEDCシステム。効率化実現を目指します。
MEDIDATAの「Rave EDC」は、試験の規模・期間・複雑さに関わらず対応できるEDCシステムです。
Viedocはスウェーデン発のEDC+ePRO機能を備えた臨床研究支援システムです。
富士通のDdworks21から、EDC機能に特化した「EDC plus」が登場しました。
知っておきたい治験・臨床研究の基礎知識
治験に関する専門知識・用語は多岐に渡ります。ですが、その中でも、例えば、治験の種類や手順、治験に関する法律(GCP等)、治験を円滑に行うことをアシストする職業(CRCやCRA等)に関する知識は、ぜひ知っておきたい基礎的な知識・用語と呼んでいいでしょう。このページでは、そうした基礎知識について、概略的に解説しますので参考にしてみてください。なお、それぞれの用語・知識について、詳しい解説ページも用意されています。
新薬の安全性や有効性を立証し、厚生労働省からの認可・承認を得ることを目的に行われる治験には、大きく分けて、4つの段階があります。最初の段階である「第1相試験」では、心身ともに健康な人を対象に、薬の安全性を確認するために行われます。続く「第2相試験」では、新薬がどれだけの有効性を持つのかを確認するために、治療のために新薬を必要とする人を対象に行われます。続く「第3相試験」では、実際の治療における投薬に近い条件で新薬の有効性・安全性を確認するために、百から数万という規模の被験者を対象に行われます。そして最後に、これらの試験の結果を検査して、厚生労働省が審査・承認を行います。
治験には、厚生労働省による新薬の認証を得るために製薬会社が医師に依頼して行う「企業治験」、医療上の必要性を背景に、企業ではなく医師が主導で行う「医師主導治験」、命に関わる重大な疾患を持つ患者のために、既存の薬が有効でない場合に、新薬の承認、および、保険適用の認可のために本来必要な期間を待たずに行う「拡大治験」、などの種類があります。
治験におけるモニタリング業務とは、GCPや治験実施計画書、各種手順書等に基づいて、治験が正しくかつ安全に行われているかどうかについて、モニタリングする業務です。このモニタリング業務は、臨床開発モニター(CRA)が行います。
似た意味を持つ単語として、混同されることも多い「治験」と「臨床試験」。新薬開発に限らず、人体を対象に行われるあらゆる治療を兼ねた試験の一切を「臨床試験」と呼ぶのに対し、「治験」は、臨床試験の内で、特に厚生労働省からの承認・認可を得るために行われる試験のことを指します。したがって、厳密には、「治験は臨床試験である」ということは出来ますが、「臨床試験は治験である」とは言えないことになります。
治験には、大きく分けて三つの段階があります。第一の段階である「第1相試験」では、新薬の安全性を確認するため、心身ともに健康な人を対象として行われます。第二の段階である「第2相試験」では、疾患の治療のために新薬を必要とする人を対象に、新薬がどれだけの有効性を持つのかを確認する目的で行われます。最後の「第3相試験」では、百から数万という規模の被験者を対象にして、より実際の治療での投薬に近い条件で新薬の有効性・安全性を確認する目的で行われます。
GCPとは、Good Clinical Practice(医薬品の臨床試験の実施の基準に関する省令)」の略であり、治験を行うにあたって関係者が遵守しなければならないルールを定めた省令の事です。このGCPは、それぞれの国によって表現こそ違えど、先進国を始めとする国々で国際的に共有されているルールです。「治験実施計画書の遵守」や「インフォームド・コンセントの徹底」などがその具体的な内容となります。
データマネジメント(DM)とは、治験において、新薬の安全性・有効性を統計的に評価するために、各種治験関連データのデータ化を行う業務です。具体的には、
- 被験者に関する症例報告書(CRF)のデータ化
- データの不足や記入ミスの洗い出し・指摘・訂正
- データの不足・記入ミスがあった場合に、製薬企業やCRA(治験モニター)に連絡
といった作業を担当します。
治験施設支援機関(SMO:Site Management Organization)とは、治験がGCD(Good Clinical Practice)に則って行われるように、治験に関する各種業務を補助する機関です。現行のGCPが平成10年に施行されて以来、治験を海外で行う製薬会社が相次ぎ「治験の空洞化現象」が起きたことを受けて、平成15年に改正GCP省令が施行され、治験施設支援機関(SMO)が誕生しました。
臨床開発モニター(CRA:Clinical Research Associate)とは、治験がGCP(Good Clinical Practice)や事前に提出された計画書に則って行われているかどうかをモニタリングしながら、治験の工程全体を管理する仕事です。似たような業務を担当する仕事として、治験コーディネーター(CRC)がありますが、治験コーディネーターは製薬会社側の立場に立つのに対して、臨床開発モニターは医療機関側の立場に立つ、という違いがあります。
治験コーディネーターとは、治験が安全かつ円滑に行われるように、被験者、担当医師、製薬会社の間で各種調整を行う仕事です。例えば、「被験者への治験内容の説明」、「被験者の心的負担を軽減するためのケアやサポート」、「被験者の来院スケジュールの管理」、「検査・投薬日の管理」、「治験業務フローの作成」、「製薬会社への各種報告」といった業務を治験コーディネーターは行います。なお、治験コーディネーターは、臨床開発モニターと異なり、医療機関側で仕事を行います。
日本国内で医薬品を販売するためには、日本国内で治験を行う必要があります。そのため、日本法人を持たない海外法人のために、「治験国内管理人」として臨床試験業務の一切を代行する組織がICCC(In country Clinical Care-taker)です。なお、ICCCは、CRO(医薬品開発業務受託機関)の内部組織となっています。
医薬品開発業務受託機関(Contract Research Organization:CRO)は、製薬会社から委託を受けて医薬品の開発に関するさまざまな業務を請け負う機関を指します。新薬や化粧品などの開発には、臨床試験も含めて莫大な時間がかかりますが、外部機関であるCROが業務を請け負うことによって期間を短縮し、より短い期間で消費者や患者さんの元へ製品が届けられるようになります。
症例報告書(Case Report Form:CRF)とは、臨床試験(治験)によって得られた被験者や患者さんのデータ、効果や副作用に関する情報などをとりまとめた報告書のことです。まとめられた情報は製薬メーカーに送られ、新薬などの開発に用いられます。臨床試験で得られた結果がわかりやすく記載されており、有害事象への対応や医師の所見を盛り込むこと、正確な記載内容と期限内に収まるように提出することが求められます。
分散化臨床試験(Decentralized Clinical Trial:DCT)は、臨床試験(治験)を受ける被験者が医療機関に来院せず、自宅やその他の場所でウェアラブル端末などを用いて臨床試験に臨む方法を指します。2011年に初めて行われ、日本国内でも新型コロナウイルス感染症の影響などでその有効性が注目されており、自動でデータを記録・送信できるため臨床試験への負担が減らせるメリットがあります。
ADRとは、Adverse Drug Reactionの略であり、日本語では「薬物有害反応」と訳され、「新薬の投薬との因果関係が疑われるあらゆる有害かつ意図されていない反応」を指します。なお、ADRの定義は国によって異なります。例えば、WHOは「有害かつ意図されない反応で、疾病の予防、診断、治療または身体的機能の修正のためにヒトに通常用いられる量で発現する作用」をADRと定義していますが、日本の場合、新薬の投薬量についての規定はADR定義に含まれません。
しばしば、ADRは「副作用(Side Effect)」と混同されますが、副反応は「あらゆる意図されていない反応」を指し示します。
PMS(Post Marketing Surveillance)とは、厚生労働省によって既に認可され、既に販売・流通している新薬について、その安全性・有効性を評価するために行われる調査のことです。治験の段階では確認されなかった効能・副作用について情報を集めることも、同様にPMSの目的の一つとなります。
PMSは、GPSP(Good Post-marketing Study Practice:製造販売後調査・試験実施基準)ならびにGVP(Good Vigilance Practice:製造販売後安全管理基準)に則った形で行われます。
ICHとは、International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Useの略であり、日本語では、医薬品規制調和国際会議」および「日米欧三極医薬品規制調和国際会議」と呼ばれています。重大な疾患を抱えている方にとって、新薬の開発は待ち遠しいものです。しかし、各国の新薬認可に関する法律間に相互性がない場合、例えば、アメリカで開発された薬が日本で使用されるためには、日本国内での承認を一から行わなければなりません。
こうした、各国の新薬認可に関する法律に相互性を持たせるためにアメリカ、日本、EU、カナダの規制当局および製薬業界団体が中心となって行われている会議がICHとなります。
GMP(Good Manufacturing Practice)とは、少量でも人体に大きな影響を与える医薬品の安全性を確保するために、医薬品の製造にあたり遵守しなければならない規則のことを指します。日本国内では、厚生労働省が「医薬品の製造管理および品質に関する規則」および「薬局等構造設備規則」として定めています。
GMPはそれぞれの国がそれぞれの基準で定めるものですが、日本、アメリカ、欧州、カナダを始めとする先進国等では、PIC/S(Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme:医薬品査察協定及び医薬品査察共同スキーム)と呼ばれるガイドラインに則った形でGMPを定めています。
新薬の開発では、治験段階での人体を対象とした試験が行われる前に、動物実験(in vivo実験)および試験管内試験(in vitro実験)を行います。こうした治験の前に行われる試験を「非臨床試験」と呼びますが、GLP(Good Laboratory Practice)とは、非臨床が安全かつ適切な仕方で行われるために守らなければならない規則のことであり、厚生労働省が省令として定めています。1960年代に世界中で社会問題となった睡眠薬サリドマイドによる薬害事故を背景に、1979年にアメリカがGLPに関する法律を整備し、それを受けて、日本では1983年にGLPに関する法律が整備されました。
SDVは「Source Document Verification」および「Source Date Verification」の略であり、直訳すれば「ドキュメント・データの直接確認」となります。治験を評価する際に、医療機関のカルテといった資料を、製薬メーカーの担当者・病院側の治験審査委員会・厚生労働省のSDV担当者等が「直接閲覧すること」がSDVとなります。
かつては、治験において症例報告書を作成する際、紙の様式を用いていましたが、近年では、電子データでデータを作成するのが一般的になりつつあります。EDC(Electronic Data Capture)とは、このように、電子データで症例報告書を作成することを可能にするためのシステムです。具体的には、治験責任医師、治験分担医師、治験スタッフなどが医療機関のPCで入力したデータをサーバに転送するための一連のシステムがEDCと呼ばれます。
CGPとは、Good Clinical Practiceの略であり、治験を安全に進めるために製薬企業や医療機関が遵守しなければならない規則を定める省令のことです。日本国内では、欧米諸国をはじめとする各国で医薬品開発における国際的なルールとして認められているガイドラインを踏まえたうえで、厚生労働省が施行しています。GCPに違反した場合は、治験の即座中止・新薬販売の中止などを含む、法的な処分が下されます。
TMFは「Trial Master File」の略であり、ICH-GCP(International Conference on Harmonisation-Good Clinical Practic)が規定する一連の治験関連文書(Essential Document)に、 一群の治験関連文書Essential Document(必須文書)と治験の実施及びデータの品質に対する評価を補助する文書から構成される一連の文書群のことです。一般的には、「治験関連文書」と呼ばれています。eTMFとは「Electronic Trial Master File」の略であり、電子データ化されたTMF のことを指しています。
治験にはメリット・デメリットがあります。今の治療とは異なる治療が受けられる、治療に必要な費用が軽減されるといったメリットがある反面、 生活に制限がかかる・副作用の可能性があるなどのデメリットもあるのです。自分にとって治験を受けることのメリット・デメリットはどのように働くか、しっかりと考えなくてはなりません。
治験では、薬の成分を含んでいないプラセボを使って、実際に試したい治験薬を飲む人とプラセボを飲む人を比較する、という試験を行います。プラセボを飲んでいることがわかってしまうと、心理的な影響が出る恐れがあるため、必ずだれにもわからないように行うのが原則です。
治験審査委員会は略して(IRB)とも呼ばれる委員会です。治験を実施する際に設置され、第三者機関として治験に問題がないかどうかを判断します。調査をした結果、問題が見つかった場合は治験の見直し・中止などを行うこともあります。治験を問題なく実施するための大きな役割を果たしている存在です。
医師主導治験においてモニタリング業務を行う上で知っておきたい、注意点や報告書作成のポイントについてまとめています。特に報告書は、IRBにて審査されるため、簡潔に・わかりやすく記載を行うことが求められます。
医師主導治験においては、万が一被験者に健康被害の保証のため、保険などを含む必要な措置を講じておかなければならない、とされています。そこで、医師主導治験における被験者補償の措置に関する情報をまとめています。
医師主導治験を実施する場合には、まず大まかな流れを把握し、全体像をつかむことが必要になります。医師主導治験の流れは大きく分けて4つの段階に分けられますが、それぞれの段階における注意点をしっかりと把握しておくことも大切です。
医師主導治験と企業治験には、さまざまな違いがあります。そこで、進め方や患者負担軽減費、治験保険など、どのような違いがあるのかを把握しておきましょう。あらかじめ違いを知っているかどうかは治験を進める上で影響してくるため、十分に理解しておくことが大切です。
治験届の内容に対して治験責任医師の変更や治験調整医師の追加、実施医療機関の追加をするときなどは、治験計画届書を提出します。中には事前届出が必要な事項もありますので、しっかりとチェックをしておきましょう。
治験届には「30日ルール」と「14日ルール」と呼ばれる2つのルールがあります。それぞれのルールの違いを知ることにより、自分たちが提出する治験届にはどちらのルールが適用されるのかがすぐにわかるようになります。
直接病院や治験会場で治験を受けるのではなく、インターネットを活用してオンライン上で行うのがバーチャル治験です。IoTの技術を活用した新しい治験の方法といえます。参加者は自宅や、自宅近くの治験会場から参加できるのが従来の治験と異なる点です。ただ、メリットだけではなくデメリットもあり、普及率が低いなどの問題もあります。
治験を行う際に取り組まなければならないのが、盲検化です。盲検化は、治験の参加者や医師、データの収集者などの無意識による思い込みが治験結果を左右してしまうことを避けるために行われます。参加者が受けている治療内容を把握している人物を限定することにより、思い込みに左右されることのない治験データの収集が可能です。
治験では、大きく分けて「ITT」「FAS」「PPS」といった3つの解析対象集団があります。それぞれ定義が異なるため、違いを理解しておくと良いでしょう。一番大きな集団となるのはITTです。ランダム化されたすべての被験者が対象となります。ITTの中から有効性のデータがない被験者を除いたFAS、その中でも特定の要件を満たしたPPSに分類されます。
治験はあらかじめ期間などを定めて行いますが、その期間を終えることなく、中断となるケースもあります。その判断のために行われるのが、中間解析です。中間解析を適切に行うことにより、試験の早期中止が可能か、必要かについて判断できるようになります。中間解析の結果はレポートを作成し、報告しなければなりません。試験終了を考慮する場合も適切な対応が必要です。
治験は一般的に大きな医療機関などで行われることになります。大学病院や公立病院のほか、規模の大きな民間病院などが代表的です。
莫大な費用をかけて行われる治験だからこそできる限り失敗を避けるため、治験を実施する医療機関は慎重に検討する必要があります。これまでの実績や治験可能な時期などについても確認しておきましょう。
治験は治験者が試験の内容や期待できる効果、副作用などについて十分に理解したうえで取り組んでいく必要があります。そこで重要とされているのが、インフォームド・コンセントです。 インフォームド・コンセントで治験者の同意を得た上で進めていかなければなりません。仮に同意のサインをした後でも心変わりがあれば、治験への参加を断ることも可能です。
国際共同治験とは、複数の国と地域が共同かつ同時進行に行う治験のことです。日本国内で行われている治験は単一国試験と呼ばれますが、国際共同治験では幅広い国が参加し、多くの治験参加者を集めて行われます。薬の副作用に関する問題点を発見しやすく、日本が参加することでドラッグラグと呼ばれる問題にも対処しやすくなります。
PETとは放射性同位元素を含んだ薬剤を用いた診断法のことを指し、糖代謝が盛んな部分に集まる性質、体内でガンマ線が放出される性質を利用して画像診断を行うことができます。近年では認知症のための薬剤開発や抗精神病薬の用量を同定するためのPET治験が行われています。PETを用いた治験は今後もさまざまな試験に利用されていくと考えられています。
治験では開発途中の薬を服用または注入し、体調の経過などをチェックします。治験が終了したあとはすぐに次の治験に移ることはできず、一定の期間を「休薬期間」としています。治験薬によっては健康状態に影響を与えるおそれがあるため、最低でも4ヶ月(副作用のリスクが少ない食品などはさらに短い)の休薬期間を設けなければなりません。
治験には多くの人が関わっています。具体的には治験依頼者、モニター、治験審査委員会、治験担当医師、治験責任医師、治験分担医師、治験コーディネーター、治験事務局、薬剤師、臨床検査技師、放射線技師、看護師などです。これらの人々が関わることにより、倫理的かつ科学的に問題がないよう安全に治験を実施しています。
治験には併用禁止薬が定められています。これは治験薬の効果を増強・減弱する恐れのある薬剤や治験薬と同様の効果を持つ薬剤です。治験参加中の患者が併用禁止薬を服用してしまった場合、副作用出現などによる健康被害を引き起こす危険性があるだけでなく、治験に関するデータが使用できなくなってしまいます。
エンドポイントとは、治験における治験薬の有効性や安全性をはかるための評価項目です。治験におけるエンドポイントは「真のエンドポイント」と「代用エンドポイント」の2種類に分けられます。真のエンドポイントは本来求めたい評価項目であり、客観性・普遍性が認められるもの。代用エンドポイントは真のエンドポイントを予測し得る暫定的な評価項目で、実際のエンドポイントとなることも多いです。
治験では治験ボランティアに謝礼金が支払われます。治験ボランティアに謝礼金を支払う目的は4つ。拘束時間に対する謝礼、予想されるリスクに対する謝礼、経済的負担に関する謝礼、制約条件による謝礼です。相場は治験のタイプにより異なり、入院タイプの治験の謝礼金の相場は1泊あたり20,000~30,000円となっています。
被験者募集会社(PRO)は、治験や臨床試験、研究などに必要な被験者を病院に代わって募集する会社です。各会社によって保持している被験者データベースや得意としている募集地域が異なります。被験者募集会社(PRO)への依頼費用には被験者謝礼と被験者募集費用の2つの要素があり、相場は治験タイプにより異なります。
2018年4月1日に施行された臨床研究法。施工後5年で見直しが規定されているため、2023年の通常国会にて改正法案が提出される見込みとなっています。明らかになっている検討項目は「革新的な医薬品などの研究開発の促進」と「研究の信頼性確保」の2項目です。臨床研究法の適用範囲に関する案も盛り込まれています。
治験を行う場合は、文書登録が必要です。治験前では、治験審査委員会の運営に関する文書や治験審査委員会の設置者が保存する記録、医療機関の治験の実施に関する手順書などが求められます。また、治験中は医療機関での治験薬の保管・管理記録、治験実施計画書からの逸脱記録などが必要で、治験後には治験の終了報告文書などが必要となっています。
治験文書管理システムは、治験業務を行うにあたり発生するさまざまな文書管理に役立つ機能が搭載されたシステムです。文書保存に関する機能や、IRB申請関連の機能が搭載されているものが多いです。導入することによって、治験文書の電子化ができるなどのメリットがありますが、数百万円単位でコストがかかるのがデメリットといえます。
試験を行う際には多くの書類を作成し、それらを適切に提出しなければなりません。「治験の依頼等に係る統一書式」では、提出や作成・管理が求められる書類について定められています。記載する際には年は西暦で書く、整理番号は各実施医療機関で必要に応じて記載するなどの注意点もあるので、事前に良く確認した上で準備が必要です。
治験薬の配送は、一般的な荷物と同様に行うことはできません。まだ世の中で流通していない薬を取り扱うことになるので、厳格な管理が必要です。温度管理が求められるケースも多いことに加え、何かトラブルがあったときのために専門的な知識を持って適切な対処ができる業者が配送を担当することが求められます。GCP省令などのルール遵守も必須です。
大人ではなく、子どもを対象とした小児治験もあります。大人に対しては有効な病気や用法・用量が明確になっている薬だったとしても、子どもの場合は適応外使用となるケースも多いです。そのため、小児治験を増やすことが求められますが、認知度の不足や安全評価が不十分であるなど、さまざまな理由によってなかなか進んでいないのが現状といえます。
治験事務局担当者はSite Management Associateを略してSMAと呼ばれることもあります。治験コーディネーターと協力し、治験がスムーズに行われるために必要になるさまざまな業務を担当する存在です。治験を実施する際にはたくさんの書類を準備しなければなりませんが、治験事務局担当者は治験関連の書類作成に関する専門的な知識を持っています。
治験は十分安全性を考慮した上で行いますが、万が一の副作用が出てしまう可能性はゼロにすることはできません。そこで、治験によって健康被害が起こってしまった場合、治験者救済のために補償を行う必要があります。対象となるのは、治験実施による因果関係が認められる健康被害のみとなります。治験保険への加入も検討した方が良いでしょう。
若い人と比較すると、高齢者に対する治験が行われるケースは少ないです。年齢を重ねるによって様々な病気を併発することがあり、それぞれで薬を飲んでいる高齢者もいるでしょう。すると、治験薬に思わぬ影響を与えてしまうこともあります。新薬開発を目指していくためには高齢者の治験も欠かせないものといえますが、様々な課題がある状況です。
基本的に、一般的な治験は妊娠中の方や授乳中の方を対象としていません。そのため、妊娠の可能性が全くない方のみが対象です。ただ、治験の中には妊娠・授乳中の女性を対象としているものもあり、そういったものに限り治験に参加できる場合があります。どのようなリスクが考えられるのかについては事前に十分な説明を行わなければなりません。
治験実施契約とは、製薬会社と医療機関の間で結ばれるもので、新薬の開発や既存の医薬品の効能・効果、安全性の確認を目指す臨床試験(治験)を行うための基本的なルールを定めたものです。治験実施契約では、適切な契約体制の形成やリスク管理などに注意しなくてはいけません。誤解を生む余地のない明確な契約作成を心掛け、安全性と公正性を維持することが求められます。
治験費用算定における課題は、業務量や市場価格に基づく根拠の不明確さや、被験者の進捗に応じた請求が不適切であることなどが挙げられます。これらの問題を克服するために、ベンチマーク型コスト算定の採用や、適正な市場価格に基づく治験費用の交渉、競争、および合意の文化の確立が不可欠です。不適切な治験費用によるリスクを回避し、業界全体の持続的な発展を支えるために不可欠な取組みといえます。
治験の空洞化とは、新しい薬の開発を行うための臨床試験(治験)が盛んに行われるはずの日本で、その治験数が減少し、また参加する人々も少なくなってしまう状況のこと。新たな医療技術や薬の開発が遅れるだけでなく、日本の医療レベル全体の向上が阻害される恐れがあります。この課題を解決する取り組みとして、治験の支援のための組織やパンフレットやWebサイトの活用などが挙げられます。
臨床研究中核病院は、高度かつ専門的な医療を提供し、臨床研究を推進するための基盤となる医療機関です。国が定める基準を満たした医療機関が認定され、症例数が多く、患者さんからの信頼も厚い特徴があります。これらの病院は、新しい医療技術や治療法の開発など、医学の進歩に不可欠な役割を果たしています。
臨床研究は患者や健康な人を対象に行われる研究の総称であり、幅広いタイプが存在します。観察研究は、特定の介入を行わずに自然の経過や因果関係を見つけ出すことに焦点を当てた研究です。介入研究は、新たな治療法や薬剤が患者にどのように影響を及ぼすかを調べるために用いられます。すべての臨床研究は、倫理的な観点を遵守し、公正な科学的手法に則って実施される必要があります。
臨床研究におけるオプトアウトとは、患者が研究への参加を拒否するために行う手続きのことを指します。通常、研究に参加する際には、その目的、方法、利益、リスク等を理解したうえで患者自身が同意すること(インフォームド・コンセント)が基本です。しかし、特定の研究では事前に同意を得ることが困難、あるいは不適切とされる場合があるため、オプトアウト制度が設けられています。
臨床研究は病気の診断、治療、予防に直結する研究であり、患者を対象にした試験や観察に基づいて行われます。一方、基礎研究は病気の根本的な原因や生命現象を解明することを目的としており、生物学、化学、物理学など幅広い分野で行われるものです。両者は異なるアプローチを取りますが、医学の進歩という共通の目標に向かって相互に関連し、貢献しています。
特定臨床研究とは、医療機関において病気の診断や治療方法の開発を目的として患者を対象に実施される研究です。医薬品や医療機器の開発とは異なり、独自の治療法の検証や医学的知見の追求に焦点をあてたものです。医療の進歩には、継続的な特定臨床研究の実施が不可欠であり、患者のQOL(生活の質)の向上に直結する成果が期待されているのです。
臨床研究における倫理性は、研究の正当性を支える不可欠な柱です。このため、研究の計画段階から、科学的根拠に基づく適切な方法論の選定、患者の安全やプライバシーの保護、そして透明性の確保に努めねばなりません。医師はいかなる状況下でも患者の利益を最優先し、害を加えないという原則を順守することが求められます。
治験は国内だけにとどまらず、海外でも行われています。さまざまな国の海外治験は日本からでも参加を申し込むことができ、外国の医療施設を訪れて新しい薬による治療や検査を受けられます。では、このような海外治験を実際に受けた場合には、国内治験とどのような点が異なっているのでしょうか。海外治験と国内治験の違いについて詳しく説明します。
治験は多くの場合、18歳以上の成人を対象に行われています。しかし、病気には子供特有のものもあるため、さまざまな症状に対応した未成年向けの治験が各地で行われています。未成年の場合は成人よりもさらに多くの条件があり、子供だけでは受けられないことから、必ず事前に保護者が治験内容を確認しなければなりません。
治験ボランティアは医療への貢献になるだけでなく、被験者にとってさまざまなメリットをもたらします。しかし、その一方でデメリットも存在するため、メリット・デメリットの両方を十分納得した上で治験に臨む必要があります。具体的にどんなメリット・デメリットがあるのかを確かめてください。
治験薬の管理では、「薬事法」及び「医薬品の臨床試験の実施の基準に関する省令」と「治験依頼者が作成した治験薬の取扱い並びに保管、管理及びそれらの記録に際して従うべき指示を記載した手順書(以下、治験薬の取扱い手順書)」に従い適切な管理を実施することが求められます。
緊急報告のための基準は大きく分けて「重篤で予測できない副作用」と「その他」に分類されます。前者では全て緊急報告の対象となり、自発報告や臨床試験や疫学研究中の副作用報告も含まれます。後者ではリスク・ベネフィット評価に大きな影響を与える情報などが考えられます。
未成年や生活保護受給者は対象外の治験が多いです。生活保護受給者が治験に参加して協力費を受け取ると、生活保護対象から外される可能性があるので注意してください。また、採血が多いため採血が苦手な人は向いていません。他には、アレルギー体質の方や外国人の血が入っている方、喫煙・飲酒をやめられない方は向いていない可能性が高いです。
ePROとは、施設で検査を行って収集するデータの他に、被験者本人から収集するデータを電子的に集約する方法のことです。具体的には、アプリを利用して健康状態を報告してもらいます。アプリのプッシュ通知でデータの取得漏れが軽減できることや、データの入力作業などスタッフの業務削減につながることなど、メリットが多く、近年重視されています。
被験者が同時期に複数の治験を受けたり、休薬期間を守らず短期間に続けて治験を受けたりする二重登録は、治験の質の信頼性を損ねるばかりではなく、被験者の健康リスクが高まってしまいます。二重登録防止の施策は、治験運用にあたり重要です。被験者の治験履歴を確認するシステムの運用やルール策定をして、二重登録を防止しましょう。
公務員の治験参加希望者をどのように対応すべきか迷うかもしれません。公務員が禁止されている副業は、労務の対価として報酬を得るものです。治験はボランティアであり、負担軽減費は報酬とはみなされないため、断る必要はありません。これまでに治験に参加して処分された事例もないことから、公務員でも被験者になってもらえます。
治験では信頼できるデータをとることが重要であり、そのためにGCPなどでさまざまな規制がされているほか、監査や査察があります。質の高いデータを得るには、質の高い治験実施計画書が必要不可欠で、手順の明確さやバイアスがかかりにくい手順にするといったことも重要なポイントです。
治験報告書とは、各治験実施計画書ごとに作成される報告書のことを言います。医薬品の承認申請を行うためには、規制当局(PMDA)にコモン・テクニカル・ドキュメントの提出が義務付けられています。ここでは、治験総括報告書の概要と確認しておきたいポイントについて詳しく解説していますので、CRO・CRAで勤務されている方は、ぜひチェックして下さい。